

 Listen to this article
Listen to this article |
Image by Gerd
Altmann in Pixabay |
It was during my final year of high school when I had an
opportunity to visit IISER, Kolkata for a Science Workshop (organised by
JBNSTS). There during one of our sessions on molecular biology, we were
introduced to a software in which we could play around with protein strands and
try to come up with ideal 3D configuration of the tertiary structure of the
protein. It was quite interesting messing around with the software, and a
professor there said that it was one of the craziest scientific challenges that
we were yet to sort out - the "Protein Folding Problem", a natural
catnip for scientists!
But, hey yo, hol' up! AI just "solved" it! Google AI offshoot DeepMind has actually made, as Nature puts it, "a gargantuan leap" in solving the Protein Folding Problem with its AlphaFold 2 model! It might as well be considered as the most important achievement in AI—ever!
The Protein Folding Problem - One of Life’s Great Mysteries
What any given protein can do depends on its unique 3D
structure.
Proteins are comprised of chains of amino acids. Our genes encode for these amino acid sequences. But just because you know the genetic recipe for a protein doesn’t mean you automatically know its shape. DNA only contains information about the sequence of amino acids - not how they fold into shape. The bigger the protein, the more difficult it is to model, because there are more interactions between amino acids to take into account.
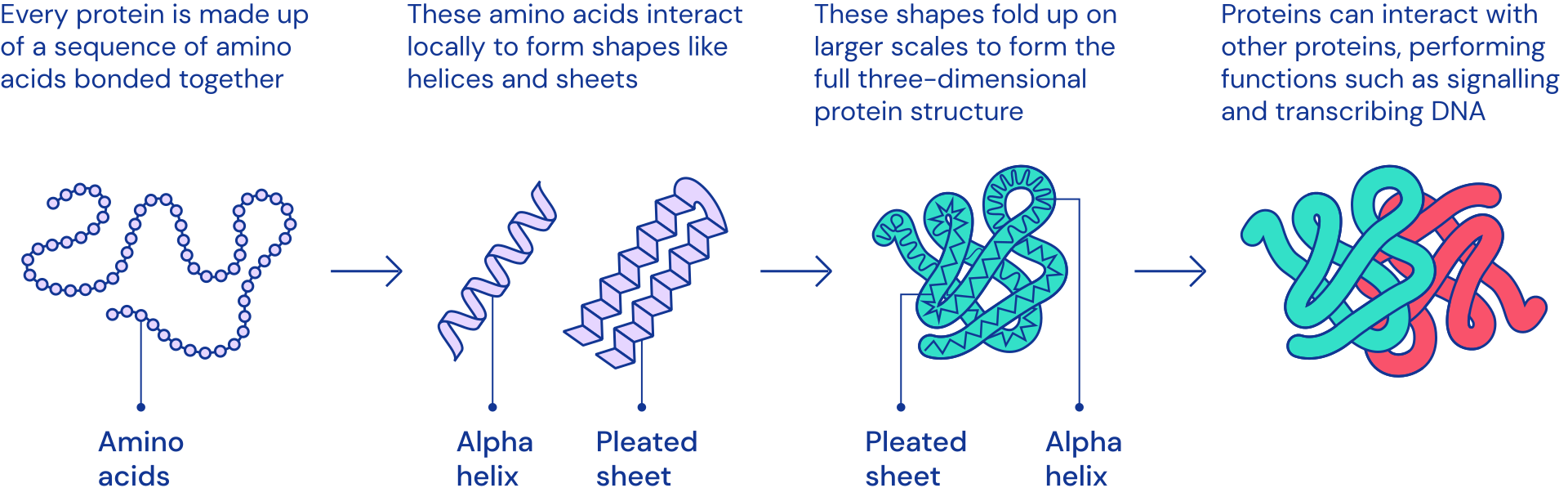 |
| Complex 3D shapes emerge from a string of amino acids. |
Basically, the protein folding problem is the question of
how a protein's amino acid sequence dictates its three-dimensional atomic
structure. The “protein folding problem” consists of three closely related
puzzles:
- What is the folding code?
- What is the folding mechanism?
- Can we predict the native structure of a protein from its amino acid sequence?
A protein’s shape is closely linked with its function, and the ability to predict this structure unlocks a greater understanding of what it does and how it works. Over the past five decades, using experimental techniques like cryo-electron microscopy, nuclear magnetic resonance and X-ray crystallography, researchers have been able to determine shapes of proteins in labs. But each of these methods depends on a lot of trial and error, which can take years of work, and cost tens or hundreds of thousands of dollars per protein structure.
In his acceptance speech for the 1972 Nobel Prize in
Chemistry, Christian Anfinsen famously postulated that, in theory, a protein’s
amino acid sequence should fully determine its structure. This hypothesis
sparked a five decade quest to be able to computationally predict a protein’s
3D structure based solely on its 1D amino acid sequence as a complementary
alternative to these expensive and time consuming experimental methods. The
ability to predict a protein’s shape computationally from its genetic code
alone – rather than determining it through costly experimentation – could help
accelerate research.
Levinthal’s paradox
The number of ways a protein could theoretically fold before
settling into its final 3D structure is astronomical. In 1969 Cyrus Levinthal
noted that it would take longer than the age of the known universe to enumerate
all possible configurations of a typical protein by brute force calculation –
Levinthal estimated 10300
possible conformations for a typical protein. Yet in nature, proteins
fold spontaneously, some within milliseconds – a dichotomy sometimes referred
to as Levinthal’s
paradox. What are the rates and routes (pathways) by which the
astronomically large conformational spaces of a protein are searched so
efficiently, by random processes, to find these uniquely structured native
states? How could order arise from disorder so fast? Pretty interesting, innit?
CASP Assessment and AlphaFold's Startling Accuracy
CASP stands for Critical Assessment of protein Structure
Prediction. In 1994, Professor John Moult and Professor Krzysztof Fidelis
founded CASP as a biennial blind assessment to catalyse research, monitor
progress, and establish the state of the art in protein structure prediction.
It is a unique global community built on shared endeavour and is also
considered the gold standard for assessing predictive techniques. Protein
structures that have only very recently been experimentally determined are
chosen to be targets for teams to test their structure prediction methods
against. Participants must blindly predict the structure of the proteins, and
these predictions are subsequently compared to the ground truth experimental
data when they become available.
According to Professor Moult, a score of
around 90 GDT is informally considered to be competitive with results obtained
from experimental methods. [GDT or Global Distance Test
is the main metric used by CASP to measure the accuracy of
predictions, and it ranges from 0-100. In simple terms, GDT can be
approximately thought of as the percentage of amino acid residues (beads in the
protein chain) within a threshold distance from the correct position.]
Surprisingly enough, in the results from the 14th CASP
assessment (2020), Deepmind's latest AlphaFold 2 system achieves a median score
of 92.4 GDT overall across all targets. In some cases, says Moult, it was not
clear whether the discrepancy between AlphaFold’s predictions and the
experimental result was a prediction error or an artefact of the experiment.
Bruh!
On its first foray into the competition, in CASP13 (2018), DeepMind with its initial version of AlphaFold, topped a table of 98 entrants, predicting the most accurate structure for 25 out of 43 proteins, compared with three out of 43 for the second placed team in the same category. That was a great surprise in itself, but the return of DeepMind with Alphafold 2 and achieving a median score of 92.4 GDT resulted in an epic "hold my beer" moment for everyone! Nobody expected such a huge leap in accuracy.
 |
| (Source: https://www.nature.com/articles/d41586-020-03348-4) |
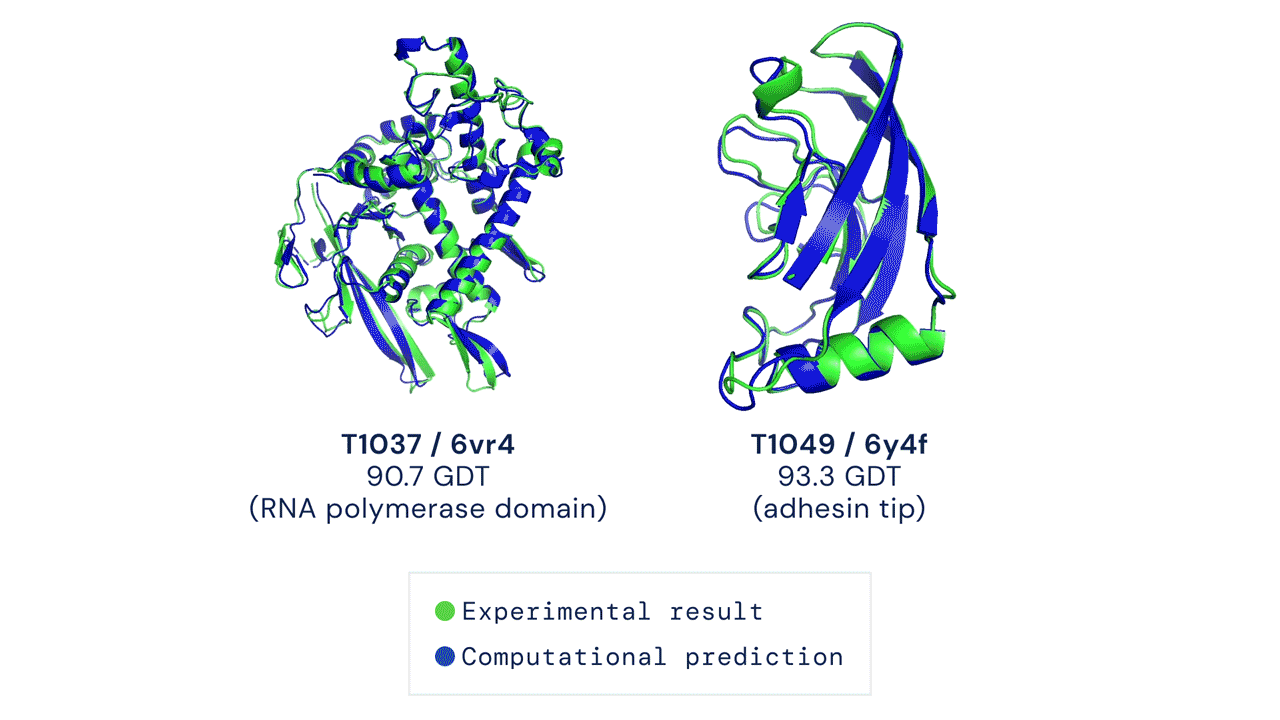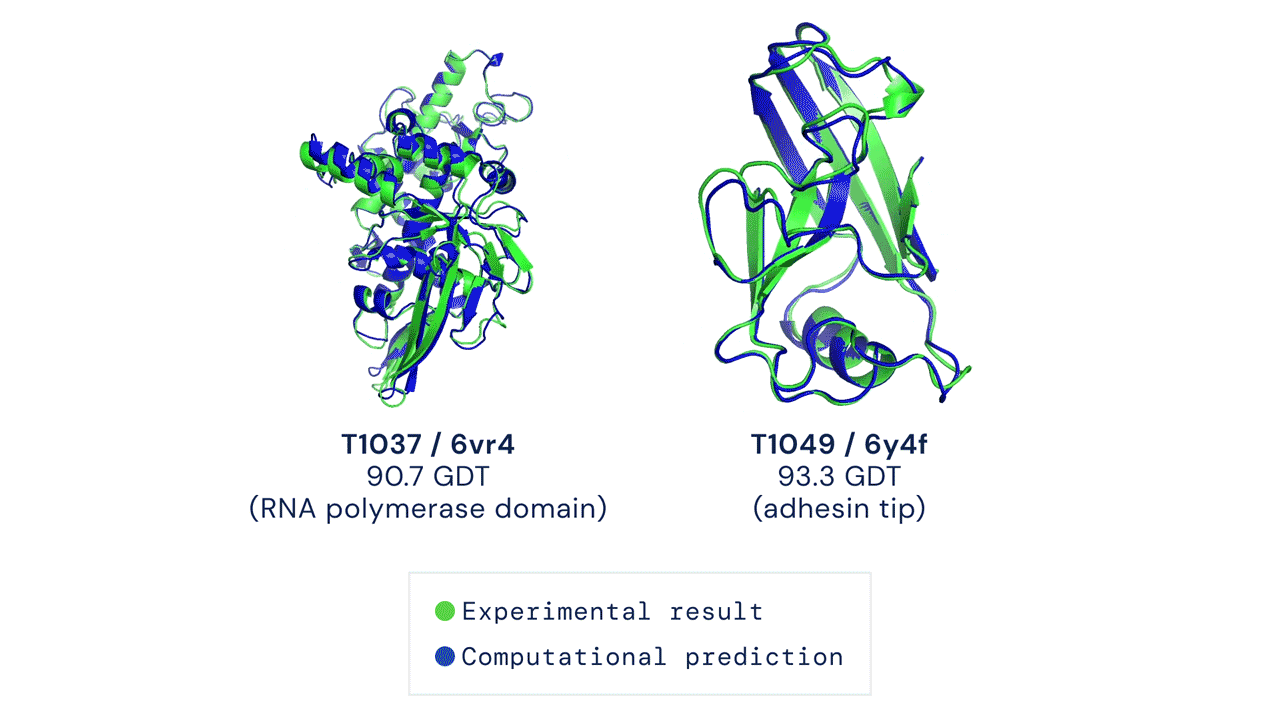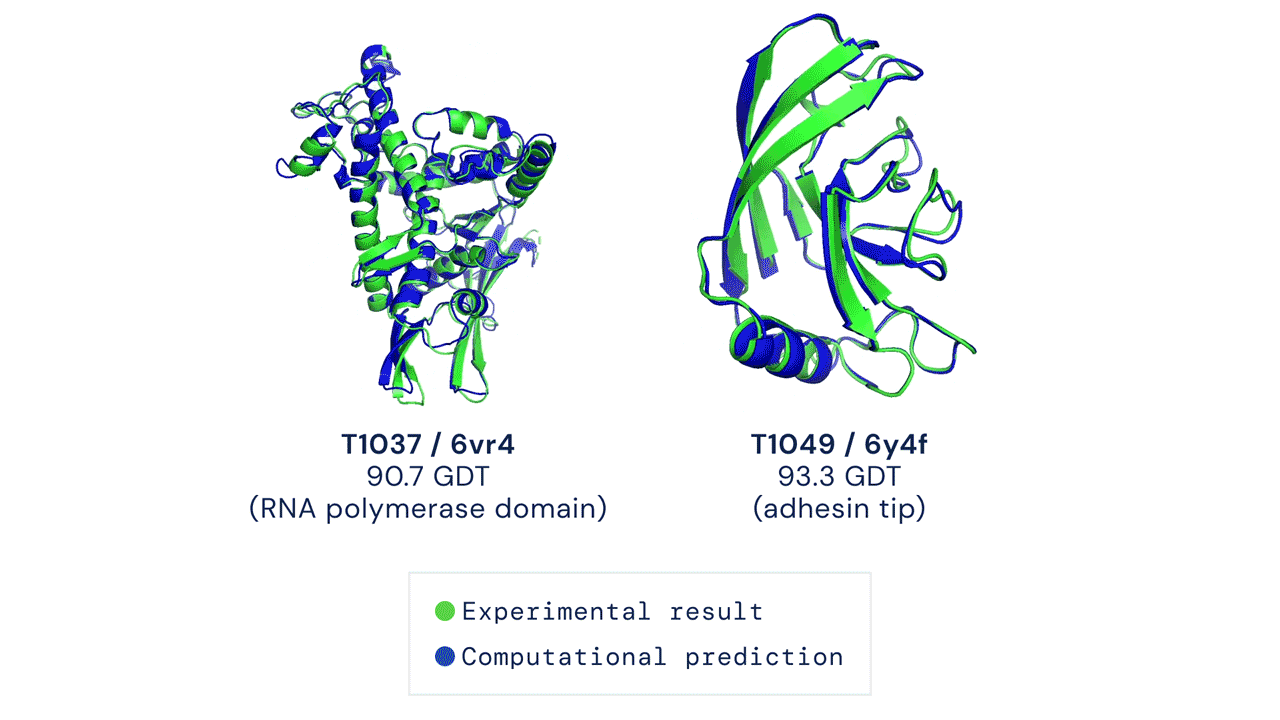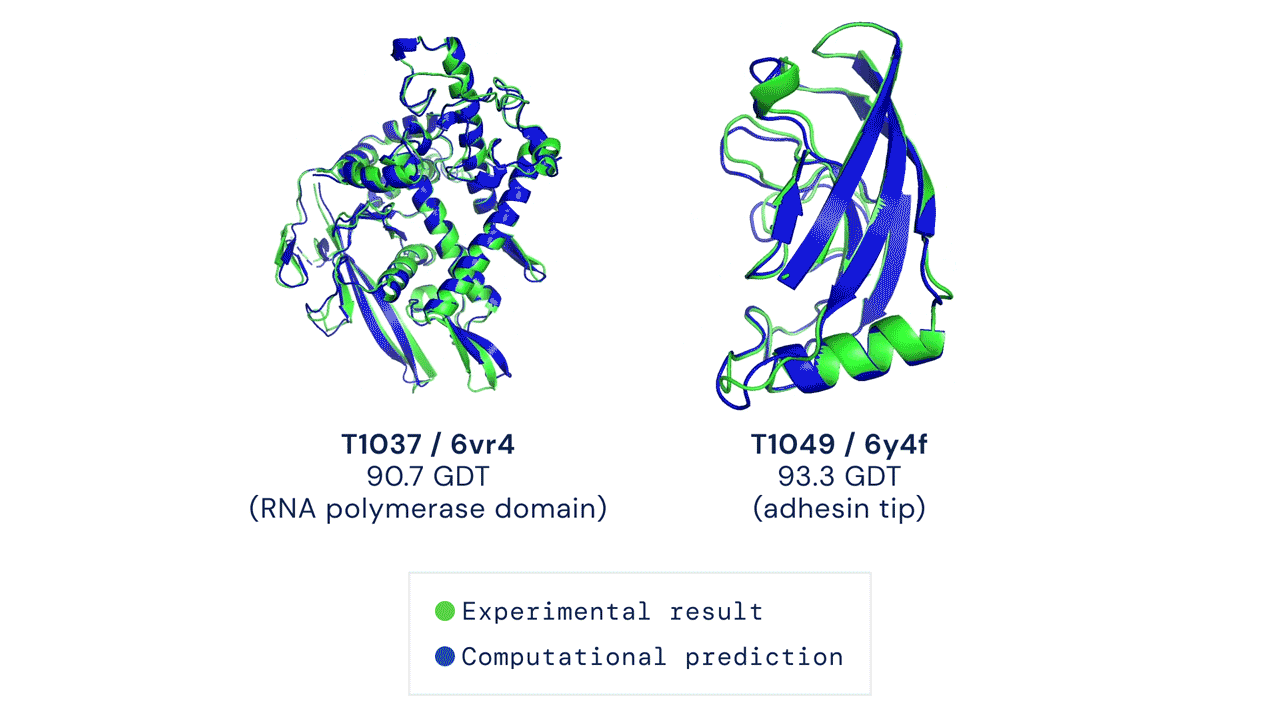 |
| Two examples of protein targets in the free modelling category. AlphaFold predicts highly accurate structures measured against experimental result. (Source: https://deepmind.com/blog/article/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology) |
The AlphaFold2 AI Model
Yep, it's complicated and it's difficult to lay down the concept in a few lines. (To dig into the details of the algorithm, you can go refer to this review article: https://web.archive.org/web/20220411065725/https://www.blopig.com/blog/2021/07/alphafold-2-is-here-whats-behind-the-structure-prediction-miracle/)
But, I would try to provide a brief overview here.
- The Training Dataset: AlphaFold was trained on a few different "open-source" (ah, one of my favorite topics that I like to talk about) or publicly available data sources. The Protein Data Bank (PDB) is a database containing the three-dimensional structures and associated amino acid sequences for virtually all proteins whose structures have been determined by mankind—around 180,000 in total, spanning human and non-human proteins. Another database, UniProt, contains the amino acid sequences (without structures) for nearly two hundred million more proteins.
- The Algorithm:
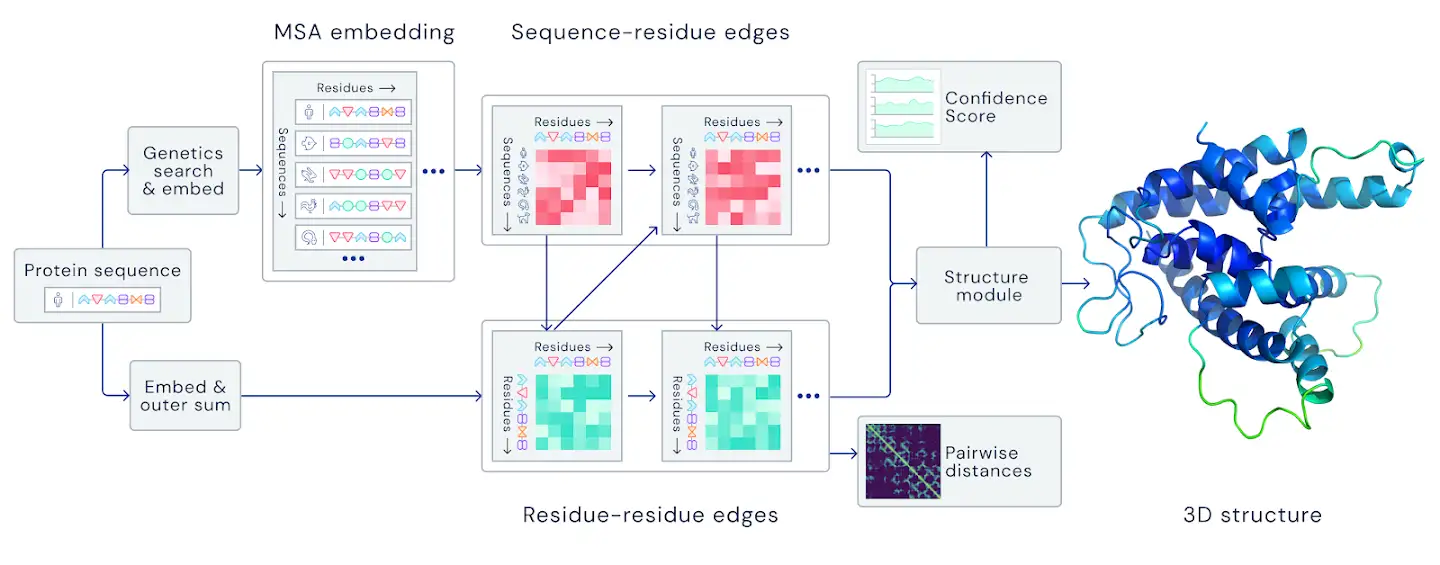 |
An overview of the main neural network model architecture. The model operates over evolutionarily related protein sequences as well as amino acid residue pairs, iteratively passing information between both representations to generate a structure. (Source: https://deepmind.com/blog/article/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology) |
It's worth mentioning that though DeepMind does have access
to far greater computing resources than the typical academic lab, AlphaFold
does not merely represent a triumph of brute-force computational power. The
amount of compute required to train AlphaFold was in fact modest relative to
other high-profile AI models. Building AlphaFold required brilliant software
engineering and several significant machine learning innovations.
In July 2021, DeepMind open-sourced AlphaFold and its
associated protein structures. This open source code provides an implementation
of the AlphaFold v2.0 system. It allows users to predict the 3-D structure of
arbitrary proteins with unprecedented accuracy. This is a move whose effects
will be felt for years to come. In the words of EMBL-EBI Director Ewan Birney:
“This will be one of the most important datasets since the mapping of the Human
Genome.”
AlphaFold 2 open-source code: https://github.com/deepmind/alphafold/
AlphaFold Protein Structure Database: https://alphafold.ebi.ac.uk/
The Potential Real-world Impact
AF2 is dope AF.👌
It represents the first time that AI has significantly
advanced the frontiers of humanity’s scientific knowledge. It has major
implications for solving many 21st-century problems, impacting on health,
ecology, the environment and basically on anything that involves living systems.
Evolutionary biologist Andrei Lupas says, “This will change
medicine. It will change research. It will change bioengineering. It will
change everything.” AlphaFold has already enabled Lupas’ lab to determine the
structure of a protein that had eluded it for a decade.
It is going to have a huge impact on drug discovery and
protein design. Knowing the three-dimensional shape of a prospective protein
target is essential to this process because a protein’s shape defines which and
how other molecules will bind to it. AlphaFold makes available a vast new set
of drug target candidates to explore.
Figuring out the most viable and impactful ways to translate
AlphaFold’s fundamental insights into products that create value in the real
world will entail years of hard work from researchers and entrepreneurs.
The European Molecular Biology Laboratory (EMBL), the
non-profit research organization in charge of stewarding AlphaFold, opines,
“AlphaFold will provide new insights and understanding of fundamental processes
related to health and disease, with applications in biotechnology, medicine,
agriculture, food science and bioengineering. It will probably take one or two
decades until the full impact of this development can be properly assessed.”
 |
Few Meaningful Limitations of AF2
AlphaFold2, essentially, predicts one stable conformation
per protein, but proteins are dynamic and may change shape as they move through
the body. Edge cases—like intrinsically
disordered proteins and unnatural
amino acids—can trip AlphaFold up. Also. its predictions are not always as
accurate as more traditional experimental methods. It generates
predictions about individual protein structures, but it sheds little light on
multiprotein complexes, protein-DNA interactions, protein-small molecule
interactions, and the like—dynamics that are essential to understand for many
biomedical use cases. As I have outlined before, like any AI system, AlphaFold
has learned to make predictions based on its training data, and hence it may
struggle to accurately predict the shapes of unusual new proteins,
including de novo protein designs not found in nature. But,
what it has already achieved is a huge accomplishment in itself and AlphaFold
is just the beginning! Now that we are starting to reap the benefits of AI in
real life applications, structural biology (and the life sciences more broadly)
will never be the same - scientific progress in these disciplines will happen
by leaps and bounds!
Conclusion
Finding a solution to the “protein folding problem” has stood as a grand challenge in the field of biology for about half a century. It has stumped generations of scientists. Hence, AlphaFold is a scientific achievement of the first order. CASP co-founder and long-time protein folding expert John Moult put the AlphaFold achievement in historical context: “This is the first time a serious scientific problem has been solved by AI.” Systems like AlphaFold demonstrate the stunning potential for AI as a tool to aid fundamental scientific discovery.